Theoretical study of ultrafast laser pulse-induced charge migration and its decoherence and complex coherence
Ultrafast charge migration is a central topic in attosecond physics and quantum control, dedicated to tracking and manipulating the femtosecond to attosecond scale motion of electrons within molecules. The research stems from the need for real-time probing of electron dynamics after photoexcitation, which is important for understanding photochemical reactions, energy transfer and information processing. In recent years, with the development of attosecond laser technology and theoretical simulations, quantum modulation of charge migration has become possible. Our group has carried out several works in this research direction as follows:
1. Adiabatic attosecond charge migration (AACM) in a linear molecule or cation such as HCCI+ means that the system has been prepared in a superposition state e.g. of the electronic ground and first excited states, corresponding to a surplus of density of valence electrons on one side which is compensated by a deficit of electron density on the other side. Subsequently, the surplus and deficit interchange such that the surplus of electron density migrates from its initial site to the opposite site, and back, periodically. The migration proceeds adiabatically on the attosecond time domain, i.e. without diabatic transitions between eigenstates. It is associated with electronic flux that mediates the charge migration. Here, we tailor a femtosecond π/2 laser pulse such that it induces AACM in the oriented model HCCI+, with maximum electronic flux. The case study shows that the flux is generated already during the laser pulse, suggesting equivalent processes during laser initiations of AACM in many or all other systems. The results are obtained by means of quantum dynamics simulation
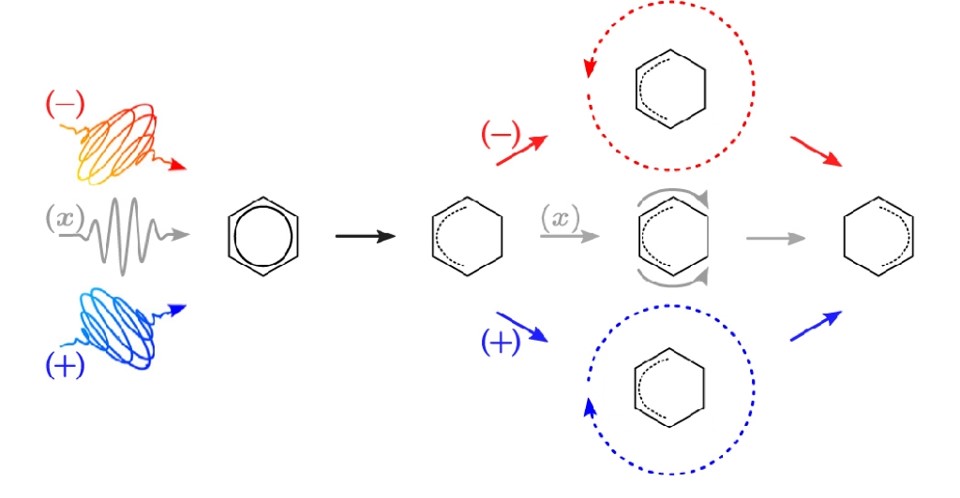
2. We design four linearly x- and y-polarized as well as circularly right (+) and left (−) polarized, resonant π/2-laser pulses that prepare the model benzene molecule in four different degenerate superposition states. These consist of equal (0.5) populations of the electronic ground state S0(1A1g) plus one of four degenerate excited states, all of them accessible by dipole-allowed transitions. Specifically, for the molecule aligned in the xy-plane, these excited states are different complex-valued linear combinations of the 1E1u,x and 1E1u,y degenerate states. As a consequence, the laser pulses induce four different types of periodic adiabatic attosecond (as) charge migrations (AACM) in benzene, all with the same period, 504 as, but with four different types of angular fluxes. One of the characteristic differences of these fluxes are the two angles for zero fluxes, which appear as the instantaneous angular positions of the “source” and “sink” of two equivalent, or nearly equivalent branches of the fluxes which flow in pincer-type patterns from one molecular site (the “source”) to the opposite one (the ”sink”). These angles of zero fluxes are either fixed at the positions of two opposite carbon nuclei in the yz-symmetry plane, or at the centers of two opposite carbon-carbon bonds in the xz-symmetry plane, or the angles of zero f luxes rotate in angular forward (+) or backward (−) directions, respectively. As a resume, our quantum model simulations demonstrate quantum control of the electronic fluxes during AACM in degenerate superposition states, in the attosecond time domain, with the laser polarization as the key knob for control.
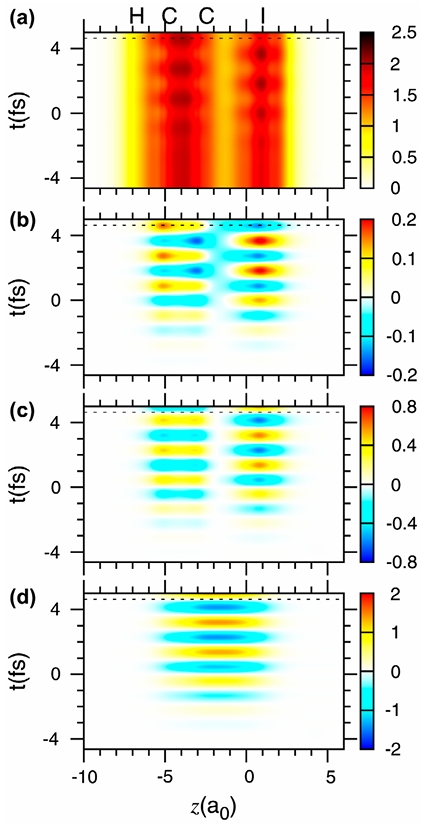
3. During charge migration, electrons flow rapidly from one site of a molecule to another, perhaps inducing subsequent processes (e.g., selective breaking of chemical bonds). The first joint experimental and theoretical preparation and measurement of the initial state and subsequent quantum dynamics simulation of charge migration for fixed nuclei was demonstrated recently for oriented, ionized iodoacetylene. Here, we present new quantum dynamics simulations for the same system with moving nuclei. They reveal the decisive role of the nuclei, i.e. they switch charge migration off (decoherence) and on (recoherence). This is a new finding in attosecond to-femtosecond chemistry and physics which opens new prospects for laser control over electronic dynamics via nuclear motions.

4.
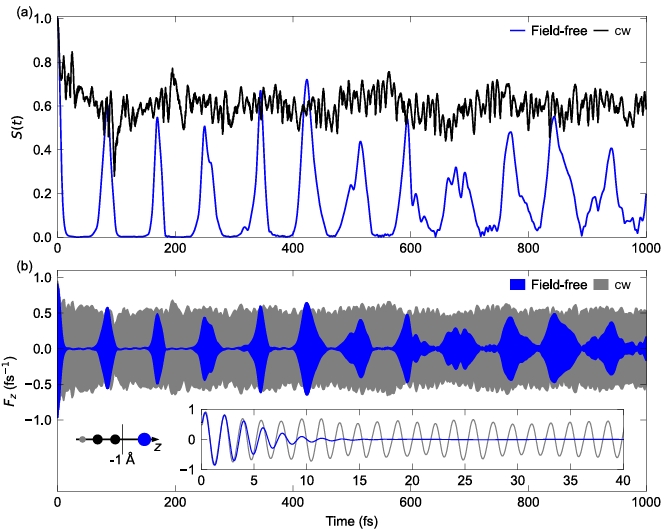
The potential applications of ultrafast charge migration in molecules have been limited by its short lifetime on the order of femtoseconds, and this rapid decay is mainly caused by nuclear motion-induced decoherence. In this paper, we propose and validate a new scheme to significantly extend the electronic coherence of charge migration using a continuous-wave laser. The scheme "anchors" the molecule with a continuous-wave laser beam that resonates with the specific electron mobility of the molecule. Theoretical studies have shown that this laser field is effective in suppressing nuclear motion, thereby significantly slowing down the decoherence process. As a result, the coherent lifetime of charge migration has been successfully extended by several orders of magnitude, resulting in picosecond and even longer "ultra-long-life" coherent evolution. This work opens up a new avenue for laser control* of electron dynamics. It demonstrates that electronic quantum coherence in molecules can be actively stabilized and maintained using a relatively simple tool such as continuous-wave lasers, which opens up new possibilities for the application of charge mobility phenomena in areas such as quantum information processing and the coherent control of chemical reactions.。